Ambiente Gráfico para o Projeto CAGE
Aluno: David Correa Martins Junior
Orientador: João Eduardo Ferreira
Parceiro de Projeto: Andrei Goldchleger
Responsável pela Disciplina: Carlos Eduardo Ferreira
Agradecimento especial a Roberto Hirata Junior e
a Carlos Hitoshi Morimoto pelas valiosas contribuições para
o desenvolvimento do projeto
Índice
1-Objetivos
1.1-Introdução
ao Projeto Cage
1.2-Problemas a resolver
1.3-Primeiras impressões
da solução
2-Metodologia de Trabalho
3-Descrição do Sistema
3.1-Escolha Geométrica
3.2-Escolha por Nomes
3.3-Scatterplot
3.4-Clustering
4-Ferramenta Utilizada
5-Atividades Realizadas
5.1-Leitura de Artigos
5.2-Reuniões
5.3-Palestras
5.4-Elaboração
de PICTIVE
5.5-Cronograma das Atividades
Realizadas
6-Considerações Finais e Resultados
7-Disciplinas Importantes
7.1-Disciplinas fundamentais
7.2-Disciplinas aplicadas
ao projeto
7.3-Disciplinas não
aplicadas, mas importantes
8-Considerações Pessoais
1.1-Introdução
ao Projeto Cage
O projeto CAGE
O projeto CAGE (Cooperation Analisys for Gene Expression) visa estudar genes através de suas expressões, utilizando uma técnica denominada Microarray ou DNA Chip. Consiste em utilizar um braço mecânico de alta precisão para depositar pequenas quantidades de cDNA em laminas de laboratório (Chips) para submetê-las a diversos experimentos. Esses experimentos normalmente são feitos utilizando substancias denominadas hibridizantes, que eventualmente reagem com os genes, produzindo alterações que posteriormente são analisadas por um microscópio e os dados resultantes são carregados para bancos de dados.
Por envolverem grande quantidades de dados e necessidade da análise
das imagens captadas pelo microscópio, o projeto necessita de forte
apoio das ciências da computação, notadamente banco
de dados e a area de visão, processamento e análise de imagens.
O IME possui dois laboratórios envolvidos no projeto: O BioInfo
- laboratório de processamento de imagens e o BioInfo - Laboratório
de banco de dados. Outros institutos que participam do projeto são
O Instituto de Química da Universidade de São Paulo e Institutos
internacionais, sendo eles: National Human Genome Research Institute (NHGRI),
NIH, Bethesda, MA, USA e Texas A&M University, Texas Center for Applied
Technology, Department of Electrical Engineering, Texas, USA
1.2-Problemas que tivemos que resolver:
Definição do Problema:
O tema central de nosso estudo foi o de desenvolver uma ferramenta computacional que permitisse que qualquer pesquisador da área do Projeto Genoma, particularmente do Projeto Cage, pudesse realizar uma busca e análise eficiente dos dados. A interface teria que ser, na medida do possível, utilizável por qualquer leigo na área de computação, mas obviamente o usuário teria que ter conhecimento do que está sendo tratado na ferramenta e como pode ajudá-lo no seu trabalho.
E um problema central que tivemos que enfrentar durante todo o projeto foi a substancial quantidade de dados que o pesquisador poderia tratar ao mesmo tempo. Isto porque o comportamento dos genes com relação a certas condições impostas é o objeto de estudo central do pesquisador da área. Por exemplo, um chip de microarray pode conter milhares de genes (até quase 20000 genes). A idéia inicial do nosso projeto é permitir que o pesquisador possa esses genes de várias maneiras.
O que descreveremos a seguir é a estrutura do conjunto dos dados que nossa ferramenta deve analisar.
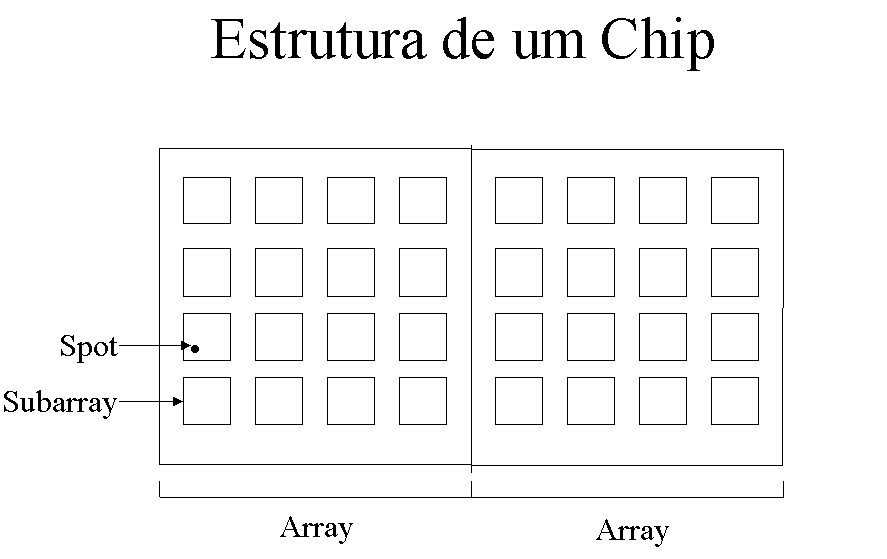
Spot hibridizado: É a menor unidade na nossa hierarquia. Cada spot hibridizado corresponde a um experimento de hibridizacao dos genes com um ou mais cdnas
Subarray: É uma unidade geométrica de agrupamento de spots. Nos dados que utilizamos para teste, um subarray e um quadrado de 22 spots de lado, totalizando 484 spots. Entretanto, outras configurações podem existir.
Array: É um grupo de subarrays. Nos dados utilizados, cada array possui 16 subarrays, totalizando 7744 spots por array.
Chip: Corresponde a uma lâmina com material genético depositado pelo braço mecânico. É composto por arrays. Nos dados utilizados, cada chip possui dois arrays, sendo que um é uma réplica do outro, para validar a medição
Subject: É a característica cuja variação está sendo estudada. Um exemplo de subject e a variação do tempo de exposição a substancias hibridizantes, com o intuito de verificar como os genes comportam-se. Normalmente, para um dado Subject, utilizam-se alguns chips, para detectar erros experimentais.
Experiment: E um conjunto de genes, que por apresentar determinada característica, torna-se objeto de estudo. Um exemplo de característica comum é que os genes pertençam a determinada linhagem, por exemplo.
Confuso??? Claro que sim, concordamos plenamente! Imagine agora a dificuldade
de criar uma interface que possa exibir todos esses dados de uma maneira
clara para que o pesquisador não se perca e ainda correlacione convenientemente
todos ou uma parte dos dados. Agora deve estar claro o nosso desafio.
1.3-Primeiras impressões da solução
Antes do nosso ingresso no projeto já havia uma pequena interface de escolha inicial dos dados. Já se havia pensado que, antes de qualquer coisa, o usuário deveria fazer um a escolha preliminar dos dados para que ele possa limitar bastante seu espaço de busca, facilitando o seu uso em atividades posteriores. Era a única parte da interface que estava mais ou menos bem definida, mas ainda apresentava certos problemas.
Foi aí então que surgiu a idéia de modificar a interface existente para exibir os dados de forma análoga ao de uma árvore de diretórios tradicional, onde o pesquisador pudesse adicionar vários conjuntos de dados da árvore para uma outra listbox. Esta interface de seleção preliminar ficou chamada de Escolha Geométrica. No tópico de Descrição do Sistema, detalharemos a interface antiga, seus problemas e a nova solução adotada.
Entretanto, com relação a outras partes do projeto, não
tínhamos muita noção de quais funcionalidades o sistema
deveria ter, como deveria ser o desenho das janelas nem a navegação
entre elas. Quando finalmente terminamos de construir a janela de escolha
geométrica, começamos a estudar como seriam as outras partes
do sistema. Mas inicialmente, só tínhamos uma solução
para a janela de escolha geométrica.
Utilizamos a programação pareada para desenvolver o sistema. O sistema foi bastante complexo de ser desenvolvido envolvendo estruturas de dados complicadas e indexação intensa de matriz. Por conta disso, tornou-se apreciável a programação pareada, pois assim que idealizávamos uma solução podíamos discutir as qualidades, seus defeitos e os problemas envolvidos ao aplicar tal solução. Na hora de implementar uma solução, ficava fácil também detectar erros que eram bastante comuns. Mesmo assim, tinha erro que nos dava bastante trabalho e tempo para ser corrigido, principalmente no início, por causa de algumas peculiaridades da linguagem utilizada.
Reuniões com o nosso coordenador também eram bastante freqüentes. Isto porque o coordenador já vivenciava o problema e a necessidade de criar um sistema utilizável para o pesquisador da área muito antes de nós ingressarmos no projeto. O processo que o pesquisador fazia para obtenção e análise dos dados já era bastante conhecida por ele. Por isso era ele quem nos orientava de maneira mais direta através de reuniões e troca de mensagens por email ou até por ICQ.
Havia reuniões esporádicas com o pessoal que cuida da parte de banco de dados do Projeto CAGE. O propósito dessas reuniões era de perguntar se havia procedimentos de bancos que faziam o que estávamos precisando. Se não houvesse tais procedimentos, explicávamos a eles nosso problema para que eles pudessem sugerir alternativas. Mas às vezes não existia alternativa a não ser encomendar os procedimentos que precisávamos para dar continuidade ao projeto.
Não havia uma especificação formal de requisitos, pois ninguém sabia muito bem as funcionalidades que o sistema deveria e poderia apresentar. Isto é devido ao fato de que este sistema tem a característica de ser algo novo e experimental. Portanto, a medida que surgiam novas idéias, discutíamos e logo implementávamos para testar a idéia. Se era satisfatório, seguíamos em frente. Se não, analisávamos os erros e adotávamos alternativas para corrigí-los.
Infelizmente, perdíamos muito tempo com esses erros, principalmente por não conseguirmos prever antes, os problemas que poderiam acontecer.
A implementação do sistema se deu exclusivamente nos laboratórios
da BIO-INFO no IME, pois era essencial a utilização do banco
de dados do laboratório paratestar trechos de código.
A interface de escolha geométrica foi o nosso ponto de partida na confecção do sistema. O objetivo da escolha geométrica é possibilitar que o pesquisador realize uma escolha preliminar dos dados com os quais deseja trabalhar, de modo a reduzir seu espaço de busca nas atividades posteriores, facilitando assim o uso. Já existia uma interface preliminar, que pode ser vista abaixo:
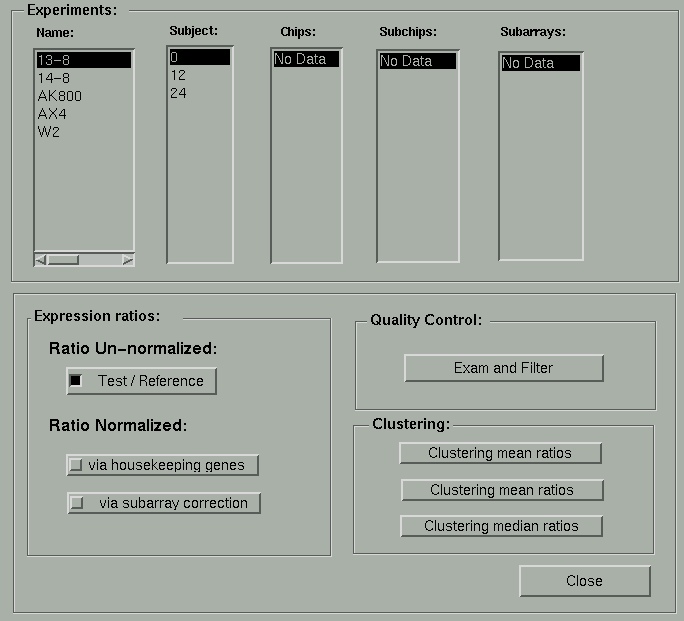
O problema com esta interface é que ela limitava a escolha dos dados a um único experimento, o que era indesejável. Partimos então para a idéia de uma interface mais flexível, que pode ser vista abaixo:
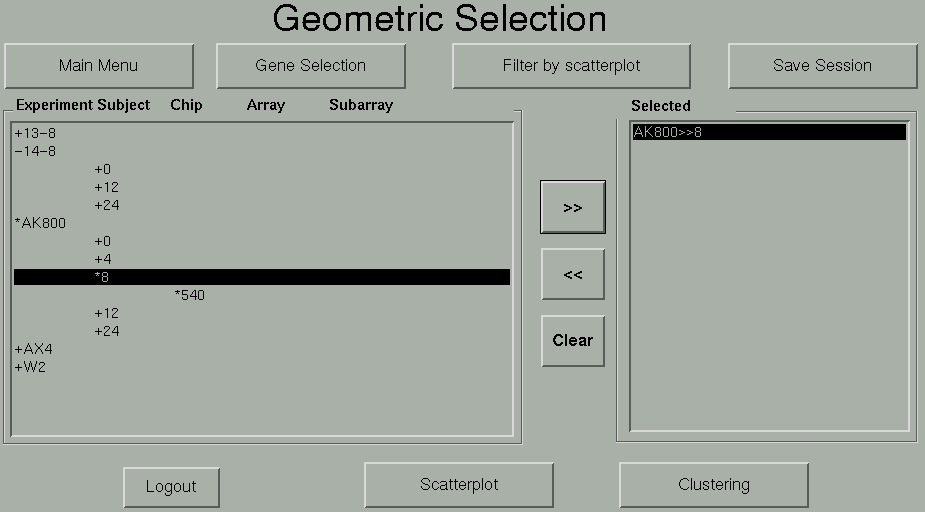
Na lista da esquerda, inicialmente aparecem os experimentos disponíveis para uso. Note os '+' antes do nome de cada experimento; isso é devido ao fato dos dados serem apresentados em uma estrutura semelhante árvore de diretórios, respeitando a hierarquia mencionada em << inserir link >>.
Clicando em um experimento, abrem-se os subjects disponiveis nesse experimento; clicando num subject, abrem-se os chips do mesmo, e assim por diante. Observe os botões etiquetados com '>>', '<<' e 'Clear'. Ao clicar em '>>' , a linha selecionada na árvore é adicionada como um caminho na lista de seleções a direita. Assim, se a linha selecionada na árvore e um chip denominado 508, do subject 12, experimento Ax4, na lista da direita é adicionado o seguinte caminho: Ax4>>12>>508. Esse caminho é marcado na árvore com '*' precedendo cada nome do caminho, e esse ramo na árvore não pode ser fechado, a não ser que o caminho seja removido da lista de seleções, clicando em '<<'. Finalmente, o botao Clear limpa a lista de seleções.
Mais um detalhe importante que definimos anteriormente sobre a escolha geométrica é que a árvore deveria exibir informações até o nível de subarray. Se a árvore exibisse informações até o nível de spot, a explosão de dados mostrada na árvore seria tão grande que se tornaria impossível para qualquer ser vivo do universo manuseá-lo sem enlouquecer. O detalhe é que mesmo até o nível de subarray, a quantidade de dados exibida ao mesmo tempo já se torna um pouco imprópria. Apesar de a árvore utilizar-se apenas de um listbox, dependendo das escolhas do pesquisador fizer, pode haver muito uso da barra de rolagem, ao contrário da interface anterior que quase não havia necessidade de usar a barra de rolagem pois cada conjunto de dados era representada numa listbox própria.
Problemas associados a essa interface:
Quando começamos a fazer a nova versão dessa interface, começamos a sentir as deficiências da ferramenta Guide do Matlab de construção de interfaces gráficas. Os Widgets disponíveis são poucos, e não existe o widget árvore. Logo, tivemos que implementar a árvore sobre um listbox comum, o que foi muito trabalhoso, pois envolvia escrever caractere a caractere na listbox, de modo a propiciar a aparência de árvore. Como na época dominávamos pouco as estruturas do Matlab, a nossa estrutura de dados também deixou um pouco a desejar, pois era a própria matriz de caracteres.
Um problema apontado pelo professor Hitoshi, em uma reunião na qual foi convidado para opinar sobre a interface, era que, conforme o usuário abre a árvore e esta se expande, cria-se a necessidade de mais espaço para visualização, o que faz aparecer a barra de rolagem. Do ponto de vista do usuário, isto é um aspecto negativo pois o excesso de scrolling necessário caso o usuário abra vários ramos de vários experimentos é grande, o que pode confundi-lo.
Assim que o usuário faz sua seleção preliminar de dados ele tem as seguintes alternativas: Filtragem por nomes de genes, Scatterplot, Filtragem por Scatterplot e Clustering.
Em qualquer uma das alternativas selecionadas, o sistema faz um pré-processamento
criando uma estrutura auxiliar para os dados selecionados na seleção
geométrica, facilitando em muitos casos a extração
dos dados em outras partes do sistema. Tal estrutura é como se fosse
um "cérebro" das operações de extração
dos dados. Por exemplo, se quisermos o chip 508 do tempo (subject) 12 do
4º experimento, a estrutura é indexada da seguinte maneira:
experiment(4).subject(12).chip(508). O problema dessa estrutura é
que ela torna-se muito esparsa consumindo muita memória. Não
descobrimos uma alternativa à essa estrutura (se a escolha do usuário
foi SOMENTE aquela mencionada acima, teremos aproximadamente 4x12x508 =
24384 posições de memória!!!). A medida que surgiam
outras dificuldades, anexávamos mais campos à essa estrutura.
Outro problema é que o sistema chegou à um ponto de ficar
praticamente ilegível para quem quiser ler o código posteriormente,
mesmo com uma boa documentação.
A escolha por nomes de genes é uma segunda opção de seleção que o usuário pode fazer com base na seleção feita na escolha geométrica. Isto permite que o usuário refine ainda mais seus dados de entrada. O sistema exibe todos os spots contidos na escolha geométrica e o usuário pode selecionar spots específicos para posterior análise. A escolha geométrica e a filtragem por nomes não servem para analisar os dados propriamente, mas são essenciais para reduzir o espaço amostral do pesquisador. Abaixo segue uma ilustração da janela de escolha por nomes.
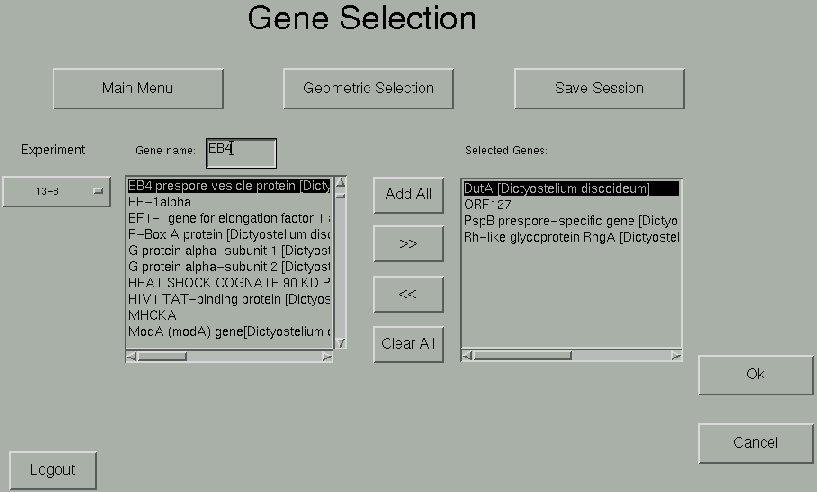
Na combo-box, sao listados os diversos experimentos que o usuário escolheu na fase de escolha geométrica. A cada experimento é associado um conjunto de nomes de genes disponíveis, resultado da escolha geométrica do usuário. Por exemplo, se o usuário escolheu apenas um chip do experimento 'Ax4', apenas os genes desse chip serão listados. Note que podem existir genes repetidos em um mesmo chip. Os genes disponíveis são mostrados na listbox da esquerda, entitulada 'Available genes' . O usuário pode realizar uma procura pelo nome do gene ou por um prefixo do nome, digitando o mesmo no campo de texto acima da listbox 'available genes'. Utilizando os botoes '>>', '<<', 'Add All' e 'Clear All', o usuário adiciona , remove, adiciona todos ou remove todos. As seleções são guardadas para cada experimento e são persistentes durante a execução do programa, isto é, o usuário pode alterar seleções já feitas sem ter que refazê-las do inicio.
Problemas associados a esta interface:
A escolha por nomes parecia uma tarefa simples, mas conforme avançávamos na implementação do sistema, encontramos dificuldades não previstas. Algumas delas foram: Necessidade de associação dos nomes de genes a cada experimento: a princípio, a interface de escolha por nomes mostrava todos os genes disponíveis no sistema, independente da seleção do usuário. Isso mostrou-se altamente indesejável, e tivemos que alterar para a forma atual, ou seja, mostramos apenas os nomes correspondentes as seleções do usuário.
Reação a mudanças nas escolhas realizadas na interface
de escolha geométrica: como o sistema permite navegação
livre, o usuário pode invalidar os nomes que ele escolheu, por exemplo,
voltando e desfazendo seleções nas quais, alguns genes estavam
incluídas na seleção por nomes.
O objetivo do "Scatterplot", também chamado de gráfico de dispersão, é determinar se certo dado sofreu alterações em seu comportamento quando submetido a condições diferentes. No caso que tratamos, o objetivo era evidenciar se os genes previamente escolhidos pelo usuário expressam-se de forma diferente quando submetidos a condições diferentes. Isto é importante pois se um gene se comportou de maneira diferente quando submetido a mudanças de condição, provavelmente tal condição é relevante para seu funcionamento, e merece estudo aprofundado.
Em nossos testes, todos os chips de todos os experimentos são idênticos em termos de distribuição de genes. As expressões gênicas podem variar, mas se em um chip, no spot identificado por x, existe o gene a, em todos os outros chips, no spot x, também existe o gene a.
A unidade de comparação dos dados em nossa ferramenta é um chip. Caso o usuário tenha selecionado partes desse chip, só estas partes serão comparadas. Entretanto, os dados devem ser consistentes, isto é, os spots de referência devem ter o mesmo identificador que os de prova. O usuário também pode escolher Subjects para realizar o scatterplot. Nesse caso, é feita uma média contendo todos os chips de um determinado subject, produzindo um chip virtual, onde o i-ésimo spot é a média das expressôes dos i-ésimos spots dos chips pertencentes ao subject.
Escolhidos os conjuntos de referência e de prova, ainda resta ao pesquisador determinar quanto devem ser as alterações entre os conjuntos para serem consideradas relevantes. No nosso caso, essas magnitudes são os fatores de subexpressão e superexpressão. Eles classificam os genes em três grupos, da seguinte forma:
Sejam:
Eref - Expressão de determinado gene
na condição de referência
Eprova - Expressão do mesmo gene na condição de
prova
Fsub - Fator se subexpressão
Fsuper - Fator se superexpressão
Se Eprova/Eref < Fsub , o gene é categorizado como subexpresso
Se Eprova/Eref > Fsuper , o gene é categorizado como superexpresso
Se Fsub >= Eprova/Eref <= Fsuper, o gene é categorizado como
não-expresso
A seguinte interface permite que o usuário realize as operações descritas acima:
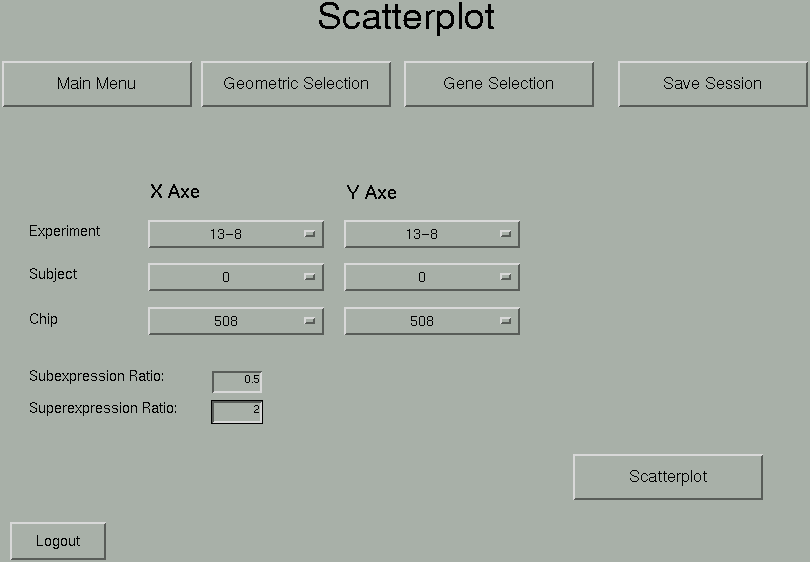
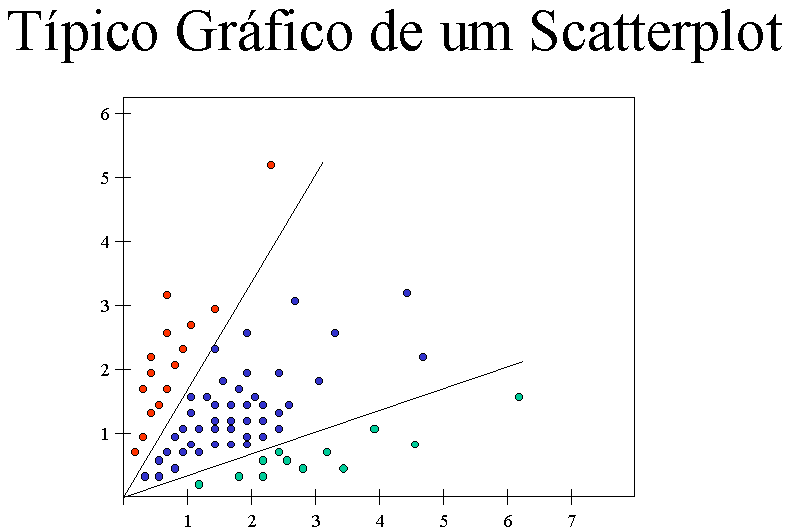
O scatterplot possui duas utilidades. A primeira é produzir um gráfico que representa o scatterplot. Nossa ferramenta gera um relatório em HTML, contendo um gráfico colorido, evidenciando cada um dos conjuntos de genes, e uma listagem dos nomes dos genes agrupados em suas respectivas categorias. Abaixo temos um exemplo de um gráfico de scatterplot:
Outra possibilidade é utilizar os conjuntos como filtragem para
atividades posteriores. Assim, o pesquisador pode decidir que, após um scatterplot, só trabalhará
com genes subexpressos, por exemplo. Tal atividade pode ser feita utilizando
a interface denominada "Filter by scatterplot", idêntica a interface
de scatterplot, com a única diferença que não gera
o relatório HTML.
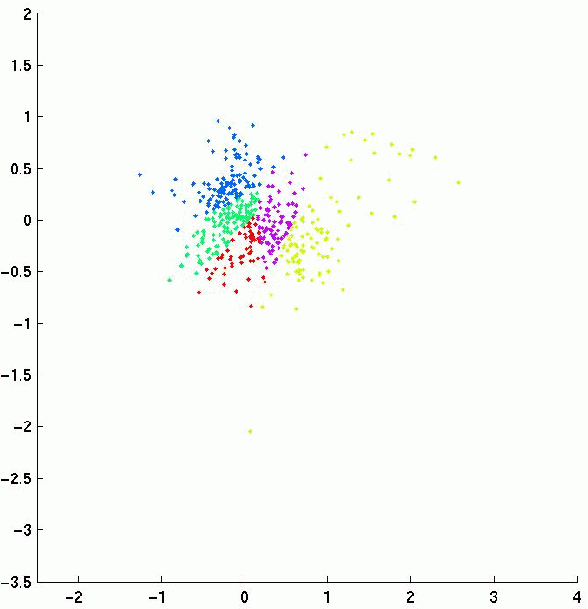
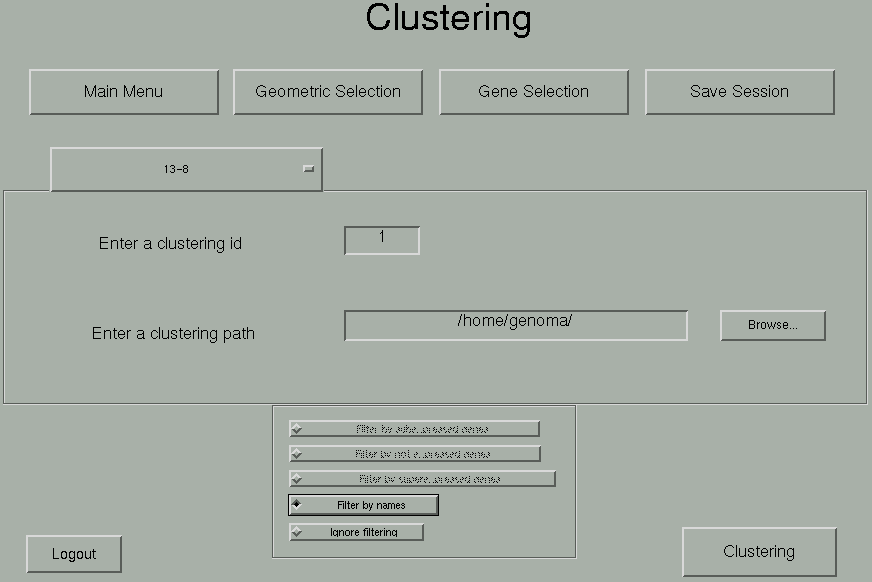
Uma outra possível análise de expressão de spots é chamada de Clustering. O Clustering é um agrupamento de expressões genéticas de acordo com um critério. O nosso sistema apenas prepara os dados para fazer o clustering, gerando automaticamente os arquivos com os dados necessários de entrada de acordo com a pré seleção do usuário e chamando a interface que realiza o clustering propriamente dito (esta parte do sistema já estava pronta antes do início de nosso projeto, não sendo necessário alterações). O clustering gera várias tabelas e gráficos. Veja um exemplo de gráfico gerado pelo clustering.
Na janela de Clustering, o usuário pode denominar um identificador ou ID do experimento em que ele deseja aplicar o clustering, escolher em qual diretório devem ser introduzidos os arquivos gerados pelo clustering, e ainda decidir se ele quer levar em conta apenas os genes escolhidos na seleção por nomes. Caso já tenha sido processado um filtragem por scatterplot dos dados, o usuário também poderá decidir se deseja trabalhar apenas com genes subexpressos, ou apenas com genes superexpressos, ou apenas genes não expressos O usuário também pode ignorar qualquer filtragem e levar em conta todos os genes incluídos na seleção geométrica feita anteriormente.
A ferramenta utilizada para criação do sistema foi a linguagem MATLAB versão 6 em conjunto com um recurso novo de criação de interfaces gráficas (GUI) para o MATLAB chamado GUIDE. Decidimos usar o MATLAB devido à maioria dos programas do laboratório terem sido escritos no MATLAB, pois o MATLAB é uma ferramenta para computação científica, própria para trabalhar com grandes volumes de dados sobre matrizes.
O GUIDE possui apenas widgets básicos para trabalhar com GUI´s. Faltava muitos recursos se comparado com outras ferramentas como o GLADE, o GTK, ou o FORTE (AWT / SWING) do JAVA. A implementação de uma árvore sobre lista, como discutimos anteriormente, foi uma atividade penosa e entediante. A falta de recursos do GUIDE nos impressionou a tal ponto que muitas vezes chegamos a achar que não era a versão completa, pois parecia que ainda estava em desenvolvimento. Mas a ferramenta já estava concluída.
O MATLAB realmente não é uma linguagem apropriada para
criação de um sistema complexo de interface orientada à
usuário, pois na hora de rechearmos um callback (codificação
de uma ação realizada pelo usuário), sentíamos
bastante dificuldades principalmente no início porque não
estávamos acostumados com as estruturas de dados internas do MATLAB.
Existia também inúmeros detalhes que demoramos para descobrir
com relação à navegação entre uma janelas,
como se podia realizar uma transmissão de dados entre janelas sem
gravação em arquivo. Sentimos também que a criação
de interfaces gráficas em um paradigma não orientado é
mais vagarosa e menos flexível do que a criação numa
linguagem com orientação a objetos.
A atividade predominante realizada durante o projeto foi a codificação
do sistema. Mas como já discutimos bastante sobre a implementação
do sistema, discutiremos aqui outras atividades pertinentes ao projeto.
Lemos alguns artigos contendo idéias sobre as técnicas que os pesquisadores utilizam para produzir as lâminas (chips) e também sobre as necessidades computacionais das mesmas, além de artigos sobre interface homem-computador, que também foi um dos principais enfoques do projeto.
Computational Analysis of Microarray Data
Autor: John Quackenbush
Neste artigo, o autor faz uma discussão sobre a real necessidade que o pesquisador do Projeto Genoma tem de ferramentas computacionais que façam cálculos estatísticos, simulações e geração de tabelas, gráficos e relatórios. Ele apresenta uma descrição sobre técnicas de coleção de dados, normalização, comparação entre expressões através de criação de gráficos estatísticos e alguns algoritmos para fazer análise de aglomerados (clustering).
A Specification Language for Direct Manipulation User Interfaces
Autor: Robert J. K. Jacob
Explicação de técnicas formais para criação
de interface orientada ao usuário final usando uma linguagem de
especificação. Utilização de diagramas de estado
(autômatos) e linguagem de programação Ada para descrever
formalmente botões, barras de rolagem, caixas de texto, popup menus,
formulários e outros widgets comuns encontrados numa interface.
Noções de orientação à objetos que podem
ser aplicados à uma interface gráfica como herança,
componentes e interação entre objetos (troca de mensagens).
Grande parte das reuniões contavam com a participação do nosso coordenador do projeto: Roberto Hirata. À medida que íamos encontrando problemas, recorríamos à ele para que nos ajudasse a solucionar esses problemas. As reuniões ocorriam com freqüência semanal.
Um dos maiores problemas que enfrentamos ao longo da implementação foi com relação a ferramenta utilizada, o MATLAB.
Mas os problemas que mais discutíamos nas reuniões não foram com relação ao MATLAB, mas sim por não sabermos o que realmente o pesquisador poderia querer. Mesmo para o pessoal da BIO-INFO, que já trabalhava há algum tempo com problemas computacionais do Projeto CAGE, era difícil saber quais funcionalidades a ferramenta deveria ter e como essas funcionalidades deveriam ser apresentadas ao usuário. Houve algumas reuniões com o pessoal do Instituo de Química para perguntar-lhes o que o sistema deveria ter, mas eles também não davam informações suficientes por do que pode e o que não pode ser feito.
Muitas vezes, nós, juntamente com o pessoal da BIO-INFO discutíamos o que o sistema deveria ter e depois verificávamos os resultados da implementação. Houve inevitavelmente muitos furos de implementação durante o projeto por não termos definido anteriormente o comportamento do sistema.
Houve reuniões com o pessoal responsável pelo banco de dados da BIO-INFO para discutirmos soluções que precisavam utilizar rotinas de banco. Além disso detectávamos muitas ineficiências em vários pontos do sistema que eram provocadas principalmente por inúmeras chamadas ao banco. Alguns desses problemas foram solucionados através da criação de rotinas de banco que trabalhassem com a maior quantidade possível de dados simultaneamente, diminuindo significativamente o overhead de conexões com o banco.
Houve também uma reunião com o professor Carlos Hitoshi
Morimoto, especialista em interfaces homem-máquina, que analisou
a nossa interface de escolha geométrica e nos deu algumas idéias
de como ela poderia ser melhorada.
Sistema de Desenvolvimento Rational Rose: Sistema que busca uma automatização da atividade de engenharia de software necessária para o desenvolvimento de qualquer tipo de sistema computacional de médio e grande porte. O nosso sistema tem um porte considerável, mas não conseguimos definir até hoje todos os requisitos necessários. Isto principalmente se deu pelo fato de que o nosso sistema possui um caráter experimental, ou seja, o sistema exibe inúmeras funcionalidades que podem ser oferecidas ao usuário final para que se possa definir o que é inútil e o que é útil. Portanto, acabamos por não optar pela utilização do sistema Rational Rose.
Palestra sobre o Banco de Dados do Projeto Genoma
Palestrante: Dr. João Eduardo Ferreira
Explicação das partes principais do banco de dados do
Projeto Genoma (Gen Bank), como se dá o relacionamento entre essas
partes com outras camadas do sistema. Divisão do banco de dados
do Projeto Genoma em duas partes principais: Projeto CAGE e Projeto Genoma
Vírus. O banco de dados do Projeto CAGE trata de dados para análise
de um único microorganismos ou célula em nível microarray
(chip, spots, etc.). Já o banco de dados do Projeto Vírus
trata de dados para análise genética sobre um conjunto de
tecidos ou um conjunto de organismos(população).
Logo depois que concluímos a janela de escolha geométrica,
tínhamos que saber como seriam as outras janelas e como elas iriam
se relacionar. O nosso coordenador nos dava informações do
que deveria ser feito dali para frente, mas nem ele e nem nós não
tínhamos idéia de qual seria uma boa forma de exibição
das informações para os dados. Tivemos então que apelar
para o PICTIVE que é uma técnica que utiliza apenas papel
e lápis para uma rápida prototipação das janelas.
Esta técnica prega que o indivíduo não perca muito
tempo no desenho das janelas (máximo 1 hora). Cada janela é
disposta em uma folha própria, inclusive as janelas de diálogo
e de erro. Quando estiver pronto, o PICTIVE permite uma simulação
do interface através de um usuário qualquer. Assim fica fácil
de detectar problemas e assimilar novas idéias com relação
à interface. E acabou dando certo, pois após o PICTIVE tivemos
idéias mais claras do que o sistema deveria apresentar e como ele
iria se comportar dadas as possíveis ações do usuário.
5.5-Cronograma das Atividades Realizadas:
Março: Reunião inicial com a equipe de banco de dados, juntamente com o Prof. João Eduardo e o coordenador Roberto Hirata. Aprendizagem inicial do recurso GUIDE de criação de interfaces do MATLAB. Entendimento do código da interface de escolha geométrica antiga e seus principais problemas.
Abril: Implementação da nova interface de escolha geométrica. Palestra sobre Banco de Dados do Projeto Genoma (Gen Bank).
Maio: Término da Implementação da interface de escolha geométrica. Criação de tabelas e estruturas de dados convenientes para utilização em todas as outras partes do sistema. Palestra sobre a ferramenta de engenharia de software Rational Rose.
Junho: Testes, correções e reformulações sobre as tabelas e estruturas de dados criadas. Entendimento das funcionalidades básicas do sistema. Elaboração de um PICTIVE para dar uma visão melhor do que o sistema deve apresentar e de como deve se comportar.
Julho: Desenho inicial no GUIDE de todas as janelas que obtivemos no PICTIVE. Reformulações das janelas.
Agosto: Inclusão de navegação entre janelas. Início da implementação da funcionalidade das janelas, começando por Scatterplot.
Setembro: Continuação da implementação do Scatterplot, testes e término da implementação do Scatterplot. Início da implementação de escolha por nome de genes. Discussão sobre trechos bastante ineficientes do código e como solucioná-los. Provamos que na maioria das vezes, as ineficiências eram causadas pelas inúmeras chamadas ao banco de dados.
Outubro: Início da implementação do Clustering. Descobertas inúmeras deficiências e inconsistências na implementação do Scatterplot, do Clustering e da seleção por nomes. Conserto da interface de Scatterplot, término da implementação de seleção por nomes e de Clustering. Testes e correções finais. Inclusão de janelas de diálogo e mensagens de erro para o usuário na tentativa de deixar o sistema à prova de qualquer usuário (user-proof).
Novembro: Término do sistema e início da monografia.
Aqui descreveremos dois objetivos que nós não conseguimos ancorar, ou por falta de tempo ou por inviabilidades decorrentes do sistema:
Integração do sistema na WEB: Era previsto desde o início que o sistema pudesse ser utilizado com relativa eficiência na Internet, por pesquisadores do mundo inteiro. Infelizmente este tópico acabou ficando inviabilizado devido a grande quantidade de dados manipulada pelo sistema. Já é relativamente lenta a execução do sistema na mesma máquina onde está instalada. Além disso, a grande quantidade de dados poderia congestionar o tráfego de dados na rede se vários usuários utilizarem o sistema ao mesmo tempo. Outro problema é que não íamos ter tempo suficiente para descobrir se haveria maneira de integrar um sistema em MATLAB com ferramentas de tecnologia WEB como Applets, Servlets ou CGI, mas pelo menos conseguimos fazer com que o scatterplot e o clustering gerassem relatórios com tabelas e gráficos em formato HTML, o que é trivial pois isto pode ser feito com qualquer linguagem de programação que permita imprimir em arquivo, sendo necessário apenas um conhecimento básico de HTML.
Uma conta para cada usuário: Esta funcionalidade foi idealizada em julho. Nessa época começávamos a vislumbrar a idéia de como seria bom se cada usuário pudesse salvar uma ou várias sessões para que posteriormente ele pudesse continuar a trabalhar exatamente da onde ele parou. Cada usuário teria um login e uma senha, onde cada um pudesse salvar quantas sessões quisesse como se cada sessão fosse uma espécie de arquivo em disco. Mas para isto eram necessárias funções e tabelas em banco de dados não previstas anteriormente. Além disso, este seria um recurso que deixaríamos por último, pois era menos importante do que outras partes do projeto, mas não sobrou tempo para a implementação dessa funcionalidade. Apesar de tudo, desenhamos as janelas de login e de menu principal associadas a esta funcionalidade, faltando o principal que era o recheio.
Apesar de não termos conseguido implementar tudo, atingimos o
objetivo principal do projeto que é o de fornecer um primeiro sistema
simples e utilizável, que seja capaz de oferecer funcionalidades
básicas que auxiliem o pesquisador do Projeto CAGE a estabelecer
relações e tirar conclusões dos dados de experimentos
envolvendo genes e suas expressões sob determinadas condições.
MAC 110 - Introdução à Computação,
MAC 122 - Princípios de Desenvolvimento de Algoritmos,
MAC 323 - Estruturas de Dados,
MAC 211 - Laboratório de Programação I
Estas disciplinas foram as que eu considero fundamentais, não
só para este projeto, mas para qualquer aplicação
da Ciência da Computação. Um curso de Ciência
da Computação sem alguma destas disciplinas não pode
ser considerado como tal.
7.2-Disciplinas aplicadas ao projeto:
MAC 446 - Interação Homem Computador: Criar um produto focado no usuário final exige que se tenha uma noção de como o usuário final se sentiria trabalhando com o sistema. Muitas vezes sentimos dificuldade em tornar o sistema utilizável devido à enorme quantidade de dados com que o pesquisador poderia trabalhar ao mesmo tempo. Em alguns pontos do sistema, o tempo de resposta é bastante lento podendo irritar o usuário. Além disso há uma série de ações possíveis que o usuário pode tomar que poderia causar erros no sistema. Tentamos tratar a maioria desses casos, mas certamente há alguns casos não tratados. Fizemos PICTIVE para entender melhor o sistema e descobrir a maioria das ações possíveis que um usuário típico poderia realizar mesmo sem ter começado a implementar o sistema.
MAC 300 - Métodos Numéricos da Álgebra Linear: Disciplina em que eu tive um primeiro contato com a linguagem MATLAB além de ter programado fazendo muitas operações sobre matrizes, o que me ajudou bastante no projeto.
MAC 239 - Métodos Formais em Linguagens de Programação: No projeto foi utilizada muita lógica envolvendo operadores AND, OR e XOR, além de ter envolvido operações sobre conjuntos como intersecção, união, união disjunta, subtração. Por isso foi importante esta disciplina, pois reduzíamos operações sobre conjuntos a simples operações lógicas.
MAC 338 - Análise de Algoritmos: Foi importante no sentido
de algumas vezes provarmos que os pontos de ineficiência encontrados
no sistema era em grande parte devido às inúmeras chamadas
a funções de banco de dados. Conseguimos reduzir um pouco
a complexidade de tempo de alguns trechos de código realizando o
mínimo de chamadas ao banco, simplesmente aplicando técnicas
de análise de algoritmos.
7.3-Disciplinas não aplicadas, mas importantes:
Aqui vou apresentar as disciplinas que não foram aplicadas, porém melhoraria substancialmente a qualidade do projeto se fossem aplicadas.
MAC 332 - Engenharia de Software: Teríamos menos dúvidas com relação à implementação do sistema se fosse feito uma análise e especificação de requisitos e funcionalidades do sistema. Talvez esta disciplina não pudesse Ter sido aplicada devido ao fato de o sistema ser algo experimental, não havendo nenhum tipo de padrão de design. Além disso, era difícil saber exatamente o que o usuário final gostaria de Ter no sistema, resultando muitas vezes em "invenções" de funcionalidades da nossa parte.
MAC 441 - Programação Orientada a Objetos: Sentimos falta do paradigma de orientação a objetos para construção do sistema, pois o MATLAB é uma linguagem procedural. Quanto maior a complexidade do sistema, maior a necessidade de orientação a objetos. O nosso código ficou inevitavelmente mal escrito, impossibilitando que alguma outra pessoa leia, entenda e adicione funcionalidades ao sistema, apesar de termos documentado razoavelmente o sistema. Não recomendo o MATLAB para construção de sistemas de médio e grande porte.
MAC 426 - Ssistemas de Banco de Dados: Apesar de trabalharmos
com chamadas de banco de dados o tempo inteiro, não sabíamos
como o banco de dados estava implementado internamente. Talvez agilizasse
o nosso trabalho se nós conhecêssemos a estrutura interna
do banco. Isto realmente era inviável pois a estrutura do banco
era bastante complexa e levaríamos muito tempo para compreendê-la.
Este projeto de iniciação científica foi uma excelente oportunidade de aprendizado das dificuldades encontradas na elaboração de um projeto de maior complexidade e como superá-las. Outra virtude de eu ter feito parte deste projeto é que de alguns anos para cá eu comecei a ficar fascinado pelos recentes resultados da engenharia genética como clonagem, cura de doenças, mapeamento genético, esforços no sentido de se descobrir a cura do câncer, entre outros.
No início deste ano eu não sabia se fazia estágio ou iniciação científica. Quando vi que muitos professores e alunos de iniciação, mestrado e doutorado estavam bastante empenhados em atividades computacionais relacionadas ao Projeto Genoma, eu pensei: por quê não unir o útil ao agradável?
Foi a partir daí que eu procurei alguma atividade computacional ligada ao Projeto Genoma. Mais particularmente, procurei algo em que eu poderia construir uma interface gráfica para auxílio às atividades do Projeto Genoma.
Então o professor João se ofereceu para ser o meu orientador de iniciação científica, pois ele estava precisando de pessoas para trabalhar na construção de uma interface gráfica para o Projeto CAGE. E aceitei sem hesitação.
E hoje estou feliz por ter me dedicado algum tempo a este projeto que caiu como uma luva para o que eu gostaria de fazer. Claro que eu senti algumas frustrações, pois às vezes o trabalho não saía do lugar, muitas vezes cheguei a pensar que este projeto não chegaria a seu fim pois descobríamos bugs em códigos feitos há um ou até dois meses atrás. Também fiquei um pouco decepcionado por não termos realizado tudo o que foi planejado no início e o que planejamos no decorrer da implementação. Ainda bem que pelo menos grande parte dos nossos objetivos foram alcançados.
A interação entre os membros da equipe foi bem direta e produtiva com reuniões semanais, respostas rápidas a eventuais dúvidas, discussão de um problema em grupo e programação pareada. Achei tal interação muito parecida com a interação entre os alunos do BCC para fazer um trabalho. Apenas uma diferença: nenhum dos trabalhos exigidos nas disciplinas do BCC tinha uma dimensão próxima a do nosso projeto que levou um pouco mais de seis meses para ser concluído. Este é mais um ponto pelo qual considerei muito importante a minha participação nesse projeto.
Planejo continuar na área acadêmica realizando outras pesquisas computacionais ligadas ao Projeto Genoma. Pretendo fazer mestrado no IME na área de Visão, Processamento de Imagens e Computação Gráfica com ênfase em aplicações da biologia molecular. Antes disso, já comecei em setembro a trabalhar na criação de uma interface Web em Servlets para coleta, armazenamento e processamento de dados clínicos e farmacêuticos do Projeto VGDN (Viral Genetic Diversity Network). Pretendo continuar com este projeto até maio e, a partir daí, começar a desenvolver um projeto de processamento de imagens relacionadas com a biologia molecular.