Estudos de técnicas de reconhecimento de padrões e aplicações em bioinformática
Orientador: Roberto Marcondes Cesar Junior
Aluno: Gláucio de Siqueira Lé
1. Introdução
As diversas aplicações de processamento de imagens e reconhecimento de padrões em problemas de bioinformática se tornaram uma das principais áreas de pesquisa do Departamento de Computação do IME-USP. Assim, o grupo de bioinformática do IME-USP tem participado do projeto temático CAGE. O orientador deste projeto participa ativamente das pesquisas em bioinformática do IME-USP, sendo que o presente plano insere-se como parte integrante desses esforços.
Ferramentas para a análise exploratória de dados estão sendo desenvolvidas de acordo com o andamento do projeto temático CAGE, o qual o grupo de bioinformática do IME-USP tem participado.
Os algoritmos declustering consistem em uma abordagem não supervisionada de reconhecimento de padrões que cumprem um papel fundamental para a análise exploratória de dados. Diante dessa importância, além do inkage, foram estudados e implementados os algoritmos particionais declustering como k-médias e fuzzy k-médias.
O grupo de bioinformática está trabalhando também em um classificador, baseado em aprendizado computacional, através de operadores morfológicos. Batizado de PAC learning system, o sistema inicialmente estava destinado a ser uma ferramenta para desenvolvimento de operadores morfológicos para imagens, mas atualmente está sendo estendido para identificação de sistemas dinâmicos e de redes genéticas. (veja http://www.vision.ime.usp.br/~nina/pac/).
Muitas vezes os problemas de reconhecimento de padrões podem demorar várias horas ou até mesmo dias para serem resolvidos. Fazendo a redução de dimensionalidade desses problemas, torna-se possível resolvê-los em minutos. Nesse contexto o bolsista vem auxiliando a extensão do sistema experimentando um método de redução de dimensionalidade no PAC Learning System.
2. Detalhes do trabalho realizado
2.1 Análise da bibliografia inicial
A leitura do capitulo 1 de [Setubal & Meidanis, 1997] proporcionou um conhecimento dos problemas atualmente estudados em bioinformática, além dos termos técnicos utilizado na biologia. Inicialmente o aluno teve uma visão geral dos métodos de reconhecimento de padrões estudando [Gose et al., 1996].
A abordagem estatística de reconhecimento de padrões é composta pelas seguintes partes: aquisição de dados (por exemplo: sensores ou câmeras); um sistema de pré-processamento, para eliminar ruídos ou distorções; um extrator de características, que cria um vetor de características com dados extraídos dos objetos adquiridos; um seletor de características, que analisa o conjunto de características e elimina as mais redundantes; e um classificador, que analisa um padrão obtido e toma uma certa decisão. O classificador toma decisões baseando-se no aprendizado obtido a partir de um conjunto de treinamento, o qual contém exemplos de padrões de todas as classes existentes no sistema. O classificador é avaliado através de um conjunto de testes, preferencialmente composto por padrões de todas as classes, mas que não estejam no conjunto de treinamento. Além do o extrator, o seletor de características podem ser dependentes dos dados de treinamento. Essa estrutura não é particular dos métodos de reconhecimento estatístico, sendo também comum em outras abordagens de reconhecimento de padrões. Mas todos os sistemas de reconhecimento possuem um extrator e/ou um seletor de atributos. Na abordagem estatística, cada padrão é representado em termos de características (features) ou atributos, sendo visto como um ponto em um espaço d-dimensional. Assume-se que cada padrão é uma observação obtida aleatoriamente segundo uma certa probabilidade condicional a uma determinada classe. Dado um conjunto de padrões de treinamento contendo elementos de todas as classes, o objetivo é estabelecer fronteiras de decisão no espaço de características que separem os padrões de classes diferentes. Na teoria de decisão estatística, a fronteira de decisão é determinada para cada classe, que pode ser especificada (aprendizado supervisionado) ou aprendida (aprendizado não-supervisionado).
Os métodos de classificação não supervisionada, como clustering, foram estudados mais profundamente a partir de [Duda & Hart, 1973] e [Jain et al, 1999].
Clustering (análise de agrupamento) é o processo de organizar uma coleção de padrões, usualmente representados por vetores de características, em grupos baseados em suas similaridades. Intuitivamente, padrões em um mesmo agrupamento são mais similares entre eles do que um padrão em um outro agrupamento. A variedade de técnicas usadas para representar os dados, medir a similaridade entre os elementos e o modo de representar os agrupamentos produziram muitos métodos de clustering. É importante ressaltar que clustering é uma abordagem não supervisionada de classificação, portanto a decisão não é baseada em um conjunto de treinamento.
Uma atenção especial foi dada para o algoritmo linkage de clustering hierárquico e para o k-médias de clustering particional, que têm sido extensivamente utilizados em análise de dados de microarray por membros do Bioinfo - USP
O linkage é um algoritmo de clustering hierárquico, ou seja, produz uma série de partições que são organizadas hierarquicamente. Esse algoritmo devolve um dendograma que representa cada partição e o nível de similaridade dos elementos agrupados.
O k-médias é um algoritmo particional, ou seja, devolve uma partição dos dados ao invés de uma estrutura de várias partições. O método particional é mais vantajoso em problemas que envolve grande volume de dados.
Além disso, o aluno estudou toda a bibliografia do curso de Visão e Processamento de Imagens, o que proporcionou uma introdução ao processamento digital de sinais, às técnicas de processamento de imagens e reconhecimento de padrões.
2.2 Procura por novas fontes de informação
Além da bibliografia indicada no projeto inicial, algumas fontes complementares têm se mostrado bastante relevantes para o amadurecimento do bolsista. Após uma busca na Internet foram encontrados vários sites sobre a tecnologia de microarray e alguns deles foram selecionados para o estudo de seu conteúdo.
Em DNA Microarray (Genome Chip) http://www.gene-chips.com foi estudado o conceito, design e as aplicações de dna microarray.
Em Division of Intramural Research - Microarray Project http://www.nhgri.nih.gov/DIR/LCG/15K/HTML/ foi estudada toda a técnica para análise de imagem de microarray, extração de informação e as atuais dificuldades encontradas no processo.
[Robert et. al, 1999] apresenta técnicas de clustering aplicadas em análise de expressões genéticas obtidas de DNA microarray. [Dougherty et. al, 2000] descreve uma utilização real de clustering para identificação de células cancerosas através de análise de expressões genéticas.
2.3 Implementação
O aluno estudou como implementar a interface para compilar rotinas escritas em C para serem executadas no ambiente matlab. Geralmente as funções em um toolbox do matlab são implementadas em forma de scripts. Esses scripts precisam ser interpretados durante a chamada de uma função, o que torna a execução mais lenta. Um programa C é compilado e não precisa ser interpretado durante a execução. É interessante também que algumas funções implementadas em C possam ser executadas no ambiente matlab para que testes e experimentos fossem mais simples de serem realizados. Basicamente, a interface que liga o programa em C com o matlab consiste de uma função (mexFunction()) que recebe os parâmetros de entrada e de saída, substituindo a função main() de um programa C comum feito para ser executado por um sistema operacional. Algumas interfaces para programas feitos por outros alunos foram implementadas para fins didáticos.
O linkage é um algoritmo de clustering hierárquico utilizado em bioinformática para análise de informação de expressões genéticas, muito utilizado pelo núcleo Bioinfo - USP. Esses fatos motivaram a implementado em C do algoritmo, para melhorar o tempo de execução, visto que a implementação existente em forma de script do matlab é bastante lenta e que deve ser utilizada muito freqüentemente em grandes volumes de dados. O algoritmo linkage está subdividido em vários outros, dentre eles, os quais foram implementados foram: Single Linkage, Complete Linkage, Average Linkage, Centroide Linkage. A diferença entre esses métodos de linkage consiste basicamente na forma de como é calculada a distância entre dois clusters, ou seja, o grau de similaridade entre os elementos de um cluster e do outro. No single linkage a distancia entre o cluster Ci e Cj é:
![]()
No complete linkage o cálculo da distância é dado por:
![]()
No average linkage o cálculo da distância é dado por:
![]()
Nesse caso ni e nj são respectivamente o numero de elementos pertencentes a Ci e a Cj
No centroide linkage a distância entre dois cluster é a distância entre os "centros de massa".
A entrada função implementada é um vetor contendo a parte triangular superior da matriz de distância. A matriz de distâncias é composta pela distância euclidiana (ou outra medida de distância) entre as amostras duas a duas. Como essa matriz é simétrica, apenas a parte superior é utilizada como entrada da função. Portanto o tamanho m da entrada é:
![]()
em que n é número de amostras.
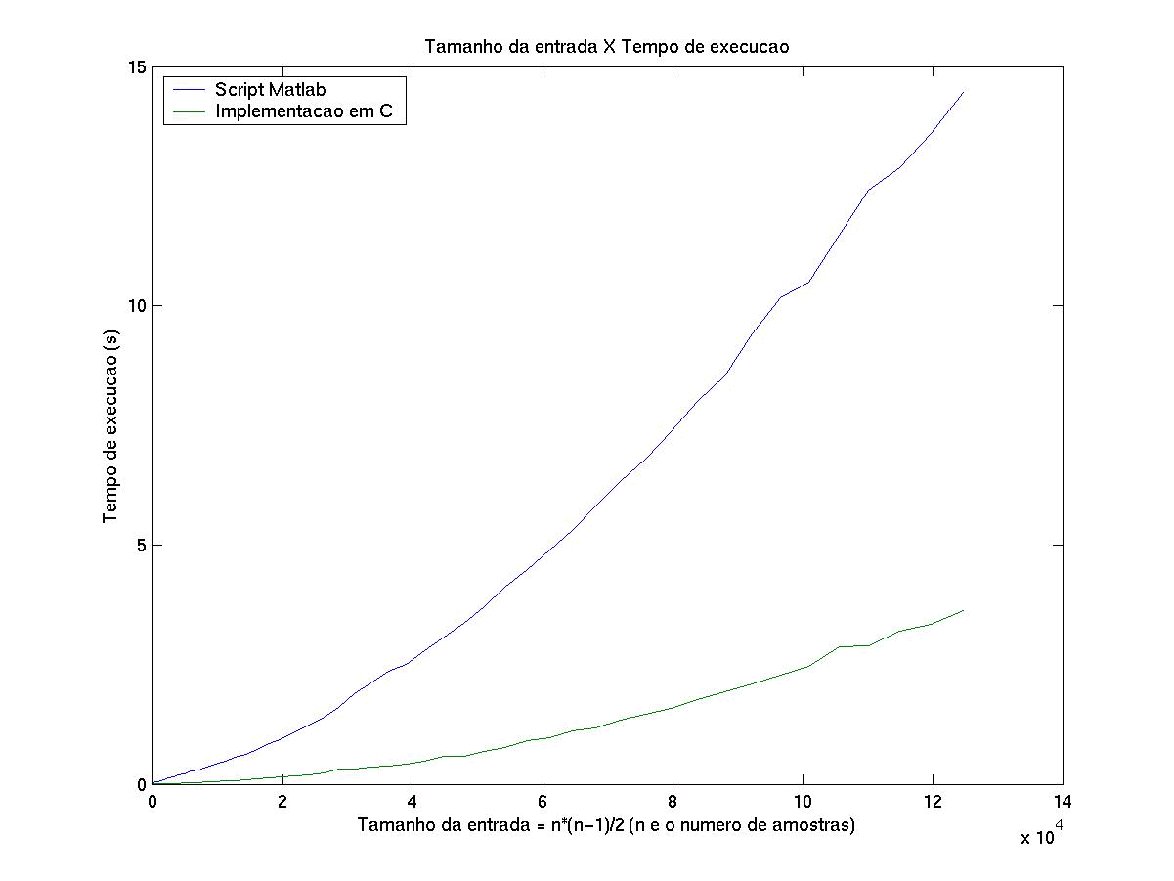
O gráfico da figura 1 representa um, teste feito com amostras aleatórias, que compara tempo de execução do algoritmo implementado em C e em forma de scripts do matlab.
 |
|
Figura 1
|
Cada amostra é um vetor de características de dimensão 3. Ambos foram testados no ambiente matab 6 para linux. Outros gráficos podem ser encontrados em http://www.vision.ime.usp.br/~glaucio/lk/index.html
A notável vantagem na performance da implementação em C sobre a implementação em forma de script deve-se ao fato de que um programa em C é compilado e não precisa ser interpretado em tempo de execução, como ocorre com os scripts do matlab. Além disso, a implementação em C é otimizada durante a compilação e foi feita de uma maneira que utiliza menos espaço temporário de memória.
O algoritmo linkage implementado em C é um clone da implementação em script Matlab. Em C o programa consumiu 311 linhas de código enquanto o script Matlab consumiu 100 linhas. Isso evidencia a facilidade de implementação que scripts Matlab proporcionam, porém essa facilidade é paga com degradação de performance, visto que a implementação em C demonstrou-se muito mais eficiente. A validação foi feita comparando os valores das saídas de ambas implementações, que foram idênticas para uma mesma entrada. (O arquivo http://www.vision.ime.usp.br/~glauco/lk/realizatestes.m é um script Matlab que executa as duas implementações do linkage, faz comparação das saídas e avalia a performance)
2.4 Implementação dos Algoritmos K-médias e fuzzy K-médias
O k-médias, é um algoritmo de clustering particional, ou seja, particiona o conjunto de dados em grupos de elementos semelhantes. O k-médias requer que o número de partições seja especificado. Um algoritmo particional como o k-médias também pode ser usado para construir uma hierarquia de partições. Aplicando novamente o k-médias em cada partição gerada, estaremos dividindo esse subconjunto em outros k subconjuntos menores, criando assim uma hierarquia de partição de maneira top-down. Esse método de criação de hierarquia contrasta com o método bottom-up do linkage por ser mais genérico, pois um grupo pode ser dividido em mais de dois grupos em apenas um passo [Gose et. al.]. O método particional é mais vantajoso em problemas que envolve grande volume de dados.
O algoritmo k-médias recebe o parâmetro k que corresponde ao número de subconjuntos em que os dados devem ser particionados. Além disso, recebe também as sementes, que são k pontos iniciais, a partir dos quais os grupos serão formados. Esses k pontos iniciais podem ser escolhidos utilizando algum conhecimento a priori dos grupos que estão querendo ser encontrados, ou podem ser escolhidos aleatoriamente.
O k-médias tenta minimizar a soma da variância inter cluster:
![]()
em que ![]() é 1 se
é 1 se ![]() pertence ao cluster k e 0 caso contrário, d
é uma função de distância e
pertence ao cluster k e 0 caso contrário, d
é uma função de distância e ![]() é o centróide do cluster k.
é o centróide do cluster k.
Os passos principais do k-médias são:
O centróide de um grupo é a média de todos elementos pertencentes a esse cluster. Inicialmente, os centróides são as próprios sementes. A proximidade é avaliada através de alguma medida de distância como a Euclidiana ou a Mahalanobis [Jain, et. al.]. O algoritmo termina quando nenhum centróide se alterou, ou seja, nenhum elemento mudou de grupo.
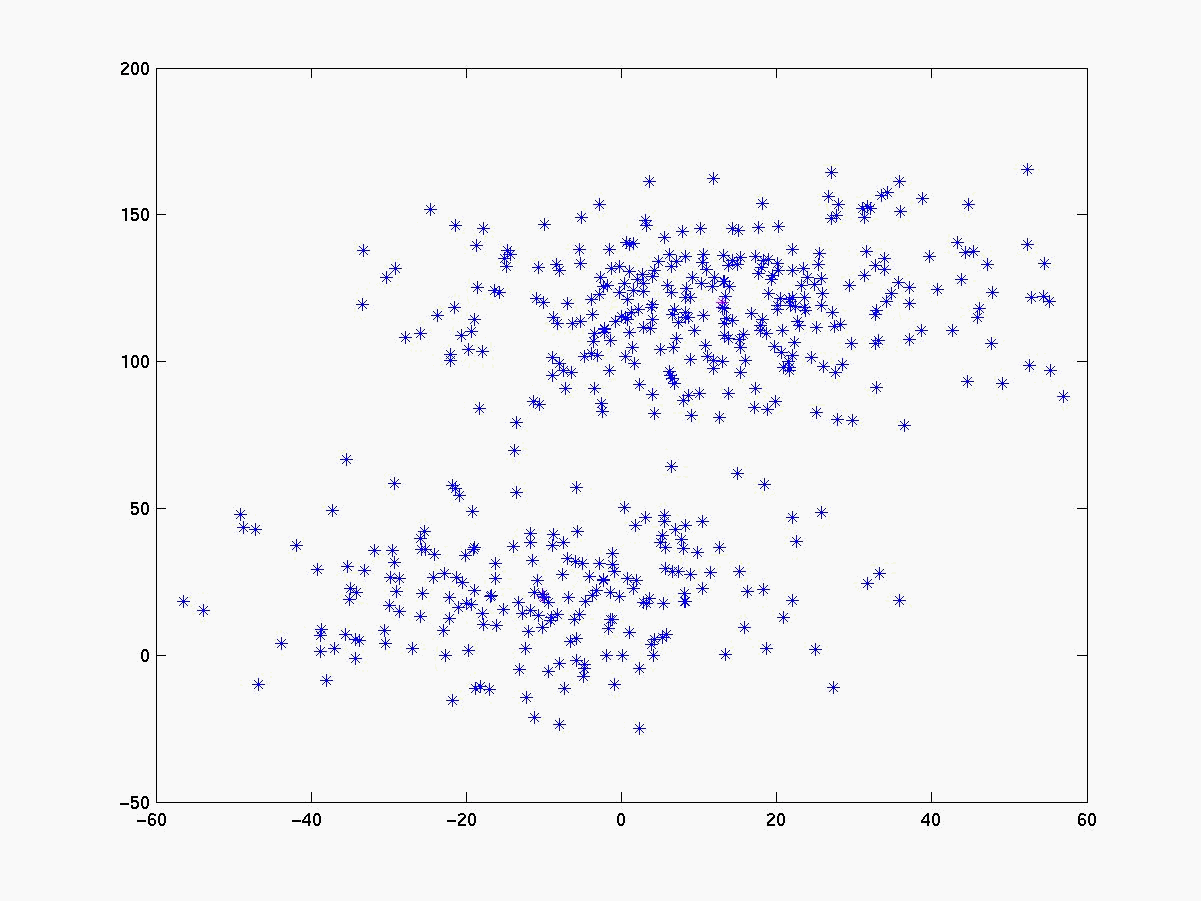
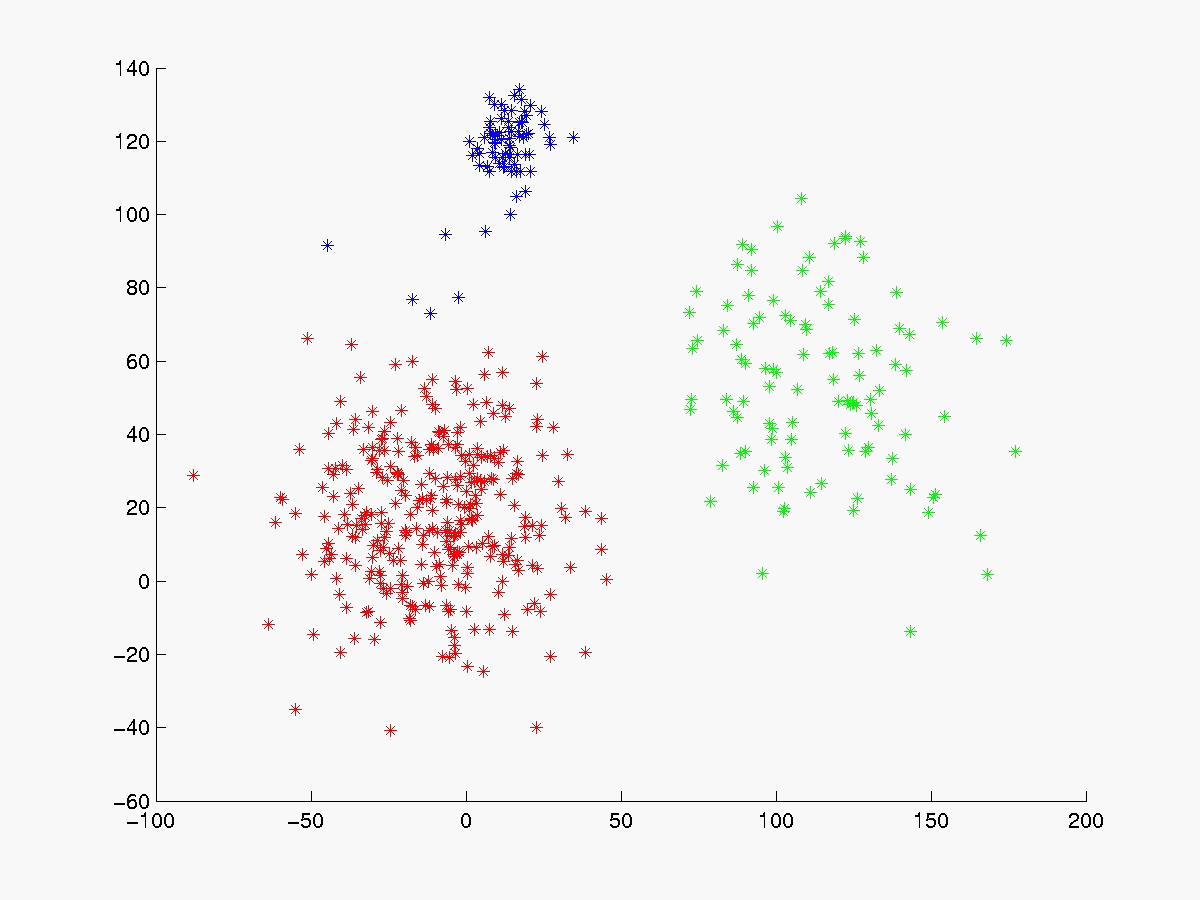
A figura 2 mostra alguns dados gerados utilizando distribuição normal [Press, 1992], enquanto a figura 3 mostra o resultado da aplicação do K-médias com k = 2
|
|
|
|
Figura 2 |
Figura 3 |
No K-médias, cada elemento pertence a apenas um cluster, portanto os cluster são disjuntos. O Fuzzy K-médias estende a noção de cluster associando cada elemento a todos os clusters através de uma função de pertinência [Bezdek, 1981]. A função de pertinência diz qual o grau de pertinência (valor em [0,1]) que cada elemento tem em cada cluster. Dessa, forma a saída desse algoritmo é um agrupamento, mas não uma partição. Em um agrupamento fuzzy, cada cluster é um conjunto fuzzy de todos os elementos.
O algoritmo tenta minimizar a função:
![]()
Em que ![]() é
o centróide do k-ésimo cluster,
é
o centróide do k-ésimo cluster, ![]() é
o grau de pertinência do elemento
é
o grau de pertinência do elemento ![]() no cluster
no cluster ![]() e
m é o coeficiente fuzzy, que pondera o quanto o grau de
pertinência influenciará na distancia d. O algoritmo implementado
recebe como parâmetro o número k de clusters, o grau de
pertinência inicial de cada elemento a cada cluster e o coeficiente
fuzzy.
e
m é o coeficiente fuzzy, que pondera o quanto o grau de
pertinência influenciará na distancia d. O algoritmo implementado
recebe como parâmetro o número k de clusters, o grau de
pertinência inicial de cada elemento a cada cluster e o coeficiente
fuzzy.
O K-médias demonstrou-se muito sensível às médias iniciais (sementes), assim como o Fuzzy K-médias foi sensível aos graus de pertinência iniciais. A porcentagem de acerto variou entre 1% a 95% durante os vários testes realizados com o mesmo conjunto de dados.
Para validação dos algoritmos implementados foram gerados clusters, com elementos aleatórios seguindo uma distribuição normal, cuja média e a variância são conhecidas. Em seguida os algoritmos foram aplicados a essas dados. A classificação de cada elemento estimada pelos algoritmos foram comparadas com a classificação conhecida.
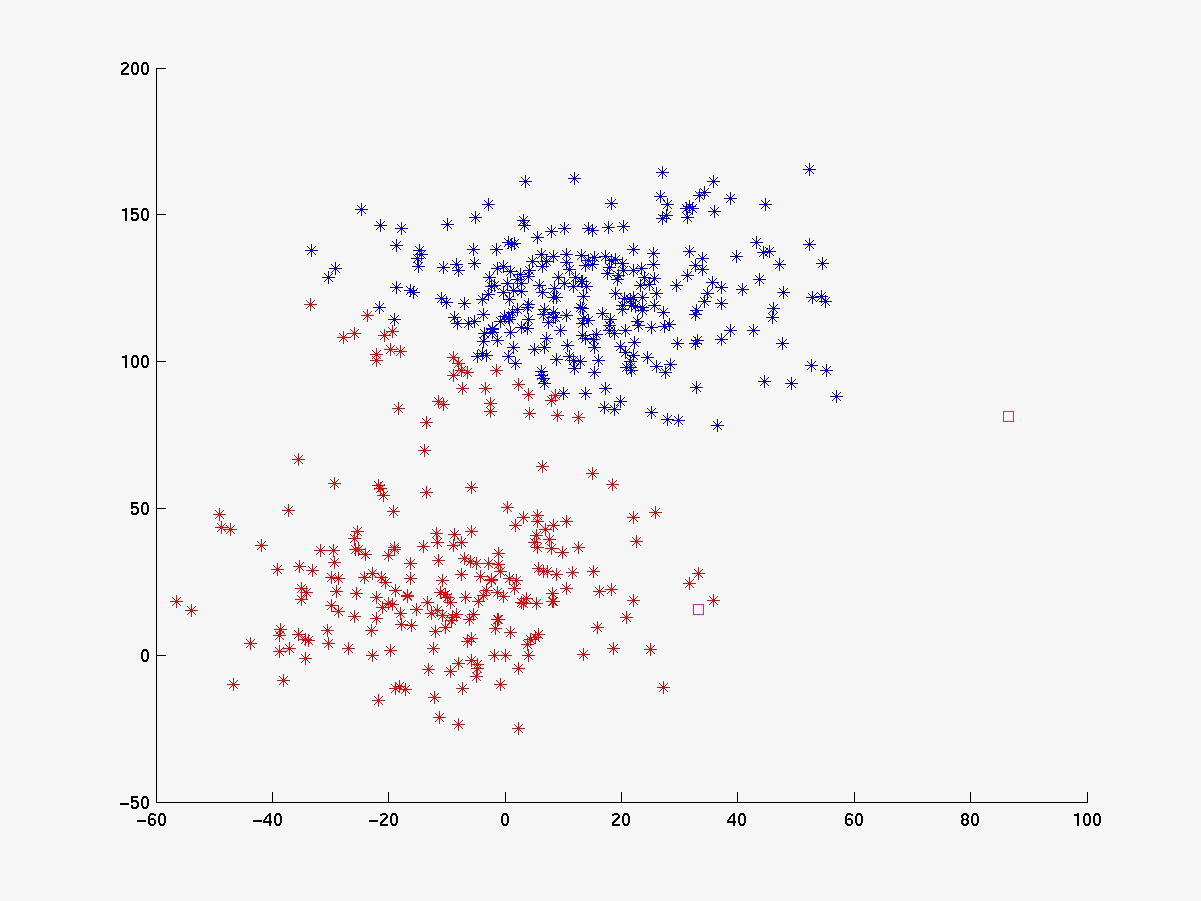
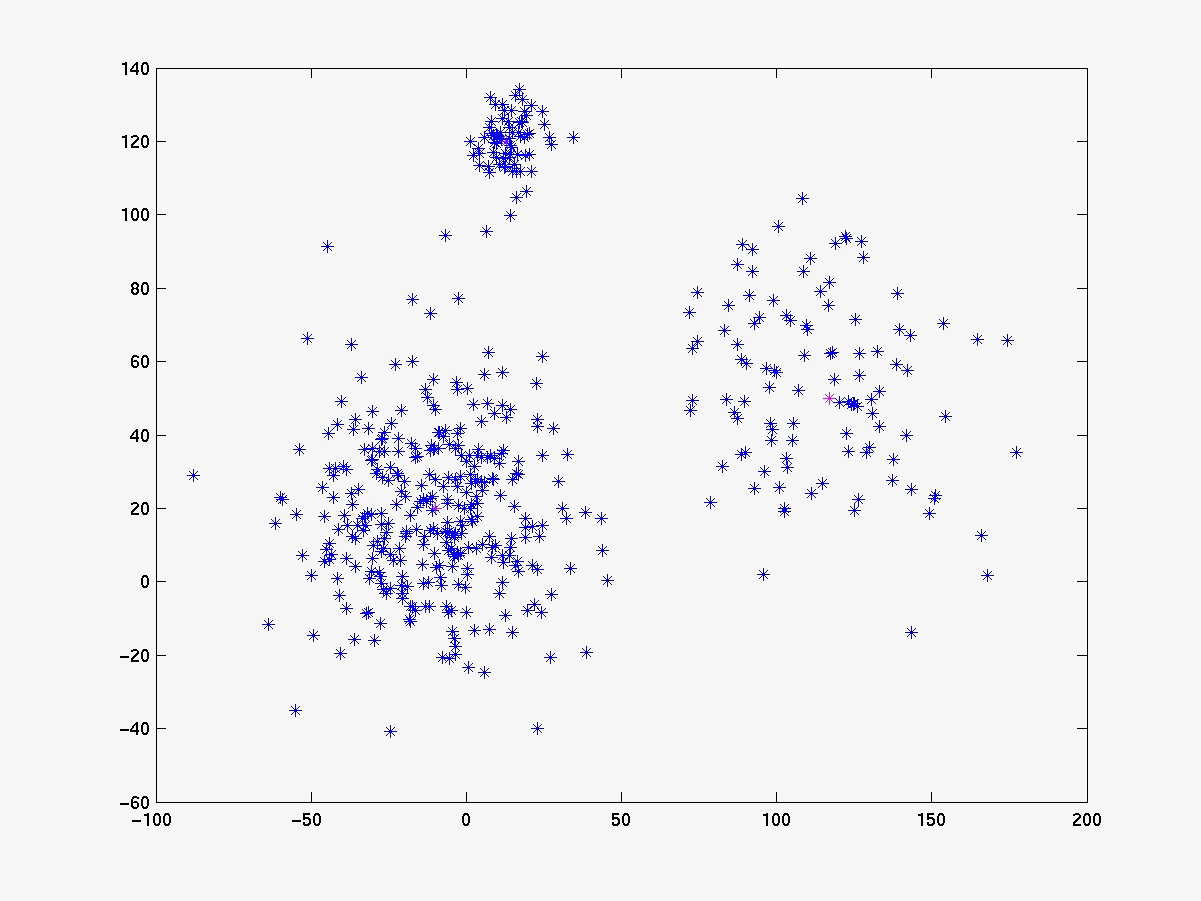
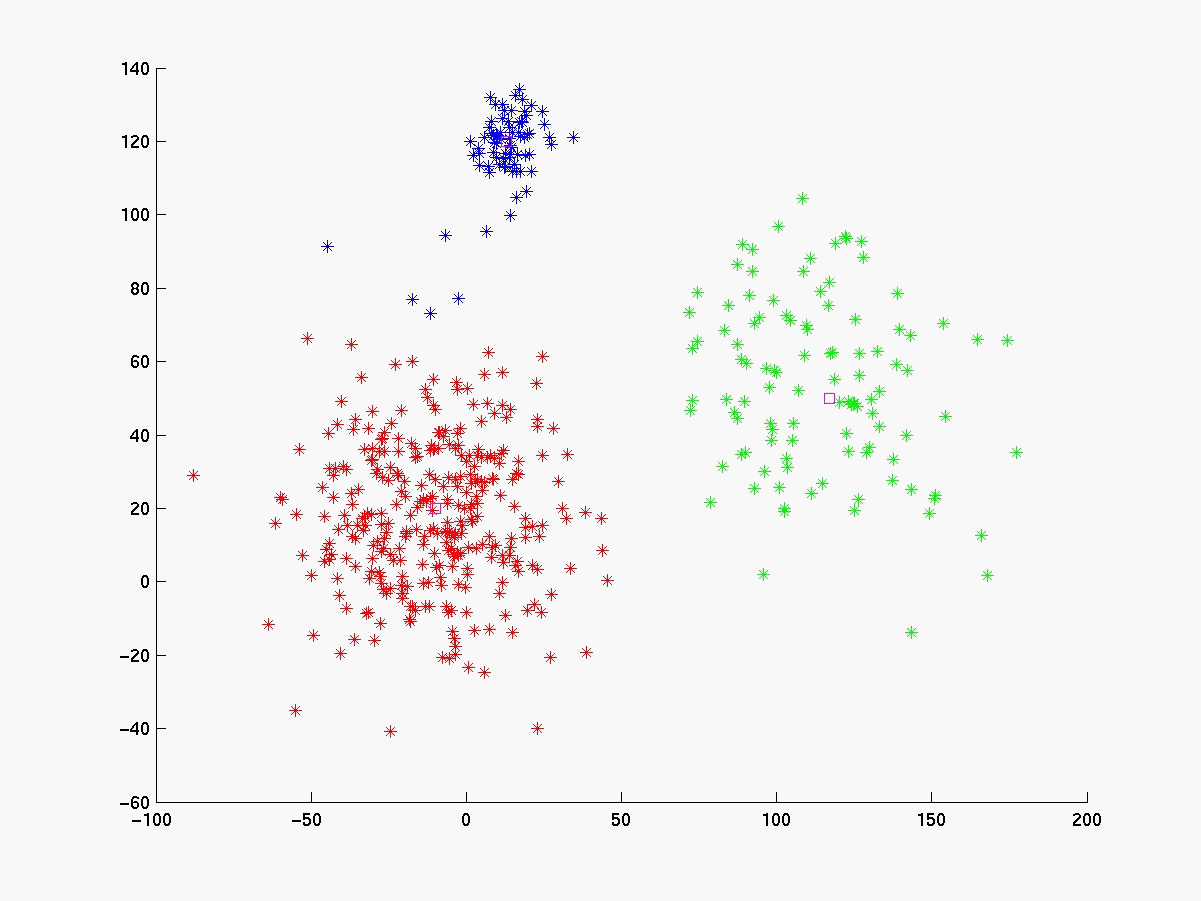
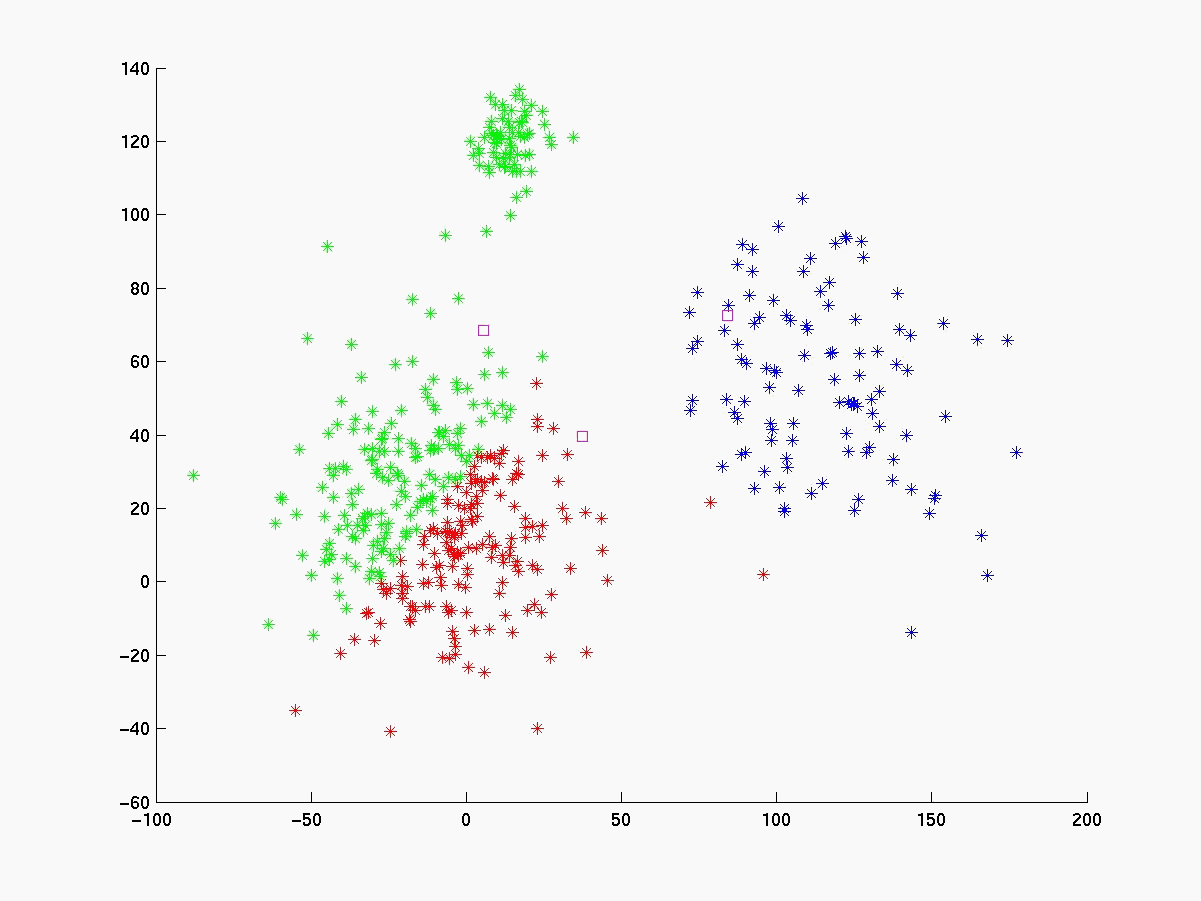
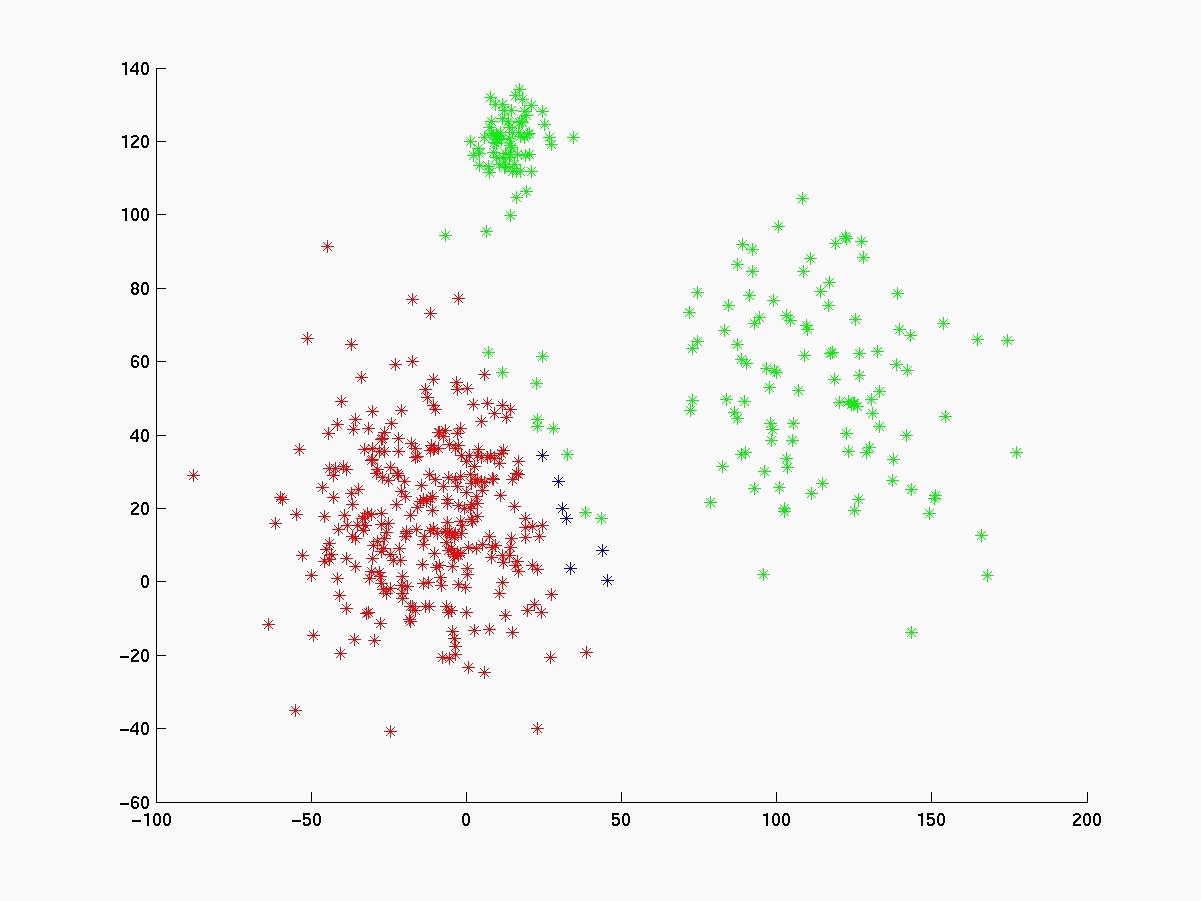
Segue abaixo a figura 4 com os gráficos dos experimentos realizados com o linkage, K-médias e fuzzy K-médias. O conjunto de dados gerado (figura 4a) está visivelmente configurado em 3 clusters cujas médias estão respresemtadas pelos pontos da cor magenta. Para esse conjunto de dados o K-médias apresentou 98% de acerto quando iniciado com as médias de cada cluster (figura 4b). Já com médias inicials aleatórias a porcentagem de acerto cai bastante (figura 4c), evidenciando sua sensibilidade às médias iniciais. O fuzzy K-médias também demonstrou-se muito sensível aos pesos iniciais (figura 4d).
|
|
|
|
Figura 4a Dados |
Figura 4b K-médias |
|
|
|
|
Figura 4c K-médias com médias iniciais aleatórias (quadrados magenta) |
Figura 4d Fuzzy K-médias com pesos inicial aleatórios. |
|
|
|
|
Figura 4e Average Linkage
|
Em conjunto de dados de 2 dimensões é fácil visualizar o resultado da classificação e verificar sua significância. Já em problemas de bioinformática, como a análise exploratória de dados de microarray, o problema torna-se complexo, pois os dados são de dimensões muito grandes. O linkage se destaca dos outros algoritmos, quando aplicado a problemas de bioinfirmática, porque não existe a sensiblidades aos parâmetros iniciais (veja figura 4e).
Para ambos os algoritmos foi implementado em C uma interface para que pudessem ser executados no ambiente Matlab 6.
2.5 PAC
O PAC Learning System é um classificador estatístico baseado em aprendizado computacional provavelmente aproximadamente correto (PAC) [Anthony, et. al.]. Essa técnica consiste em gerar uma estimação de operador ótimo (que, no caso estudado, são funções booleanas) para um dado conceito alvo. O PAC Learning System foi implementado para operar com imagens mas atualmente está sendo estendido para redes genéticas e sistemas dinâmicos.
A questão da redução de dimensionalidade
[Campos, 2001] está relacionada com o número de características,
que nesse caso é o número de variáveis, que serão
utilizadas para treinar o operador. Esse conjunto de variáveis é
chamado de janela, sendo portanto o número de variáveis equivalente
ao tamanho da janela. Como no caso estudado as função são
booleanas, elas residem em um espaço de tamanho ![]() ,
em que n é o número de variáveis ou tamanho
da janela. Pode-se mostrar que nesse espaço de operadores existe um operador
ótimo [Barrera, et. al.]. O algoritmo de aprendizado PAC retorna uma
estimação desse operador ótimo. O chamado fenômeno
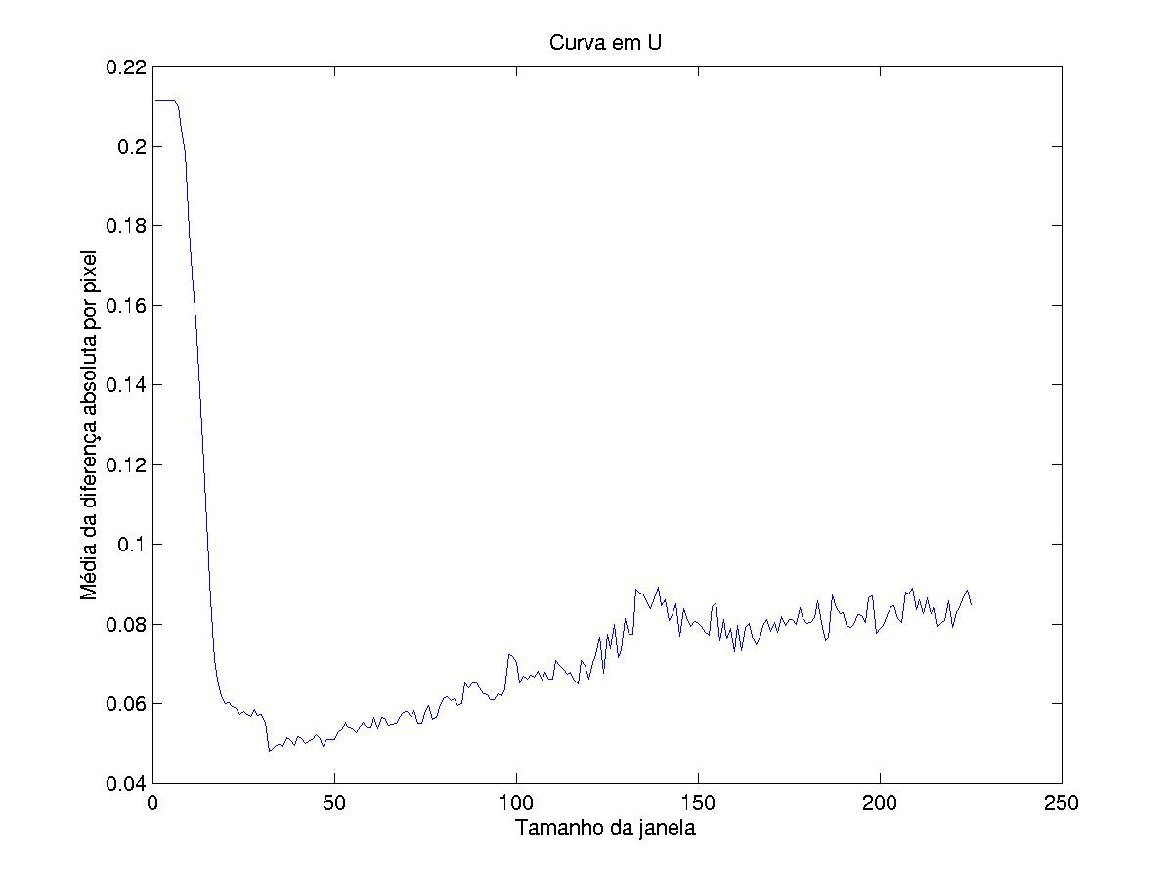
da curva em U [Campos, 2001] explica que uma janela pequena cobre uma parte
muito pequena do espaço, enquanto uma janela muito grande aumenta o erro
de estimação, estando ambos extremos associados a maiores erros.
Portanto, existe um tamanho de janela ótimo. A figura 5 é a curva
em U (gráfico tamanho da janela X erro do operador) que demostra a existência
de um tamanho de janela ótimo.
,
em que n é o número de variáveis ou tamanho
da janela. Pode-se mostrar que nesse espaço de operadores existe um operador
ótimo [Barrera, et. al.]. O algoritmo de aprendizado PAC retorna uma
estimação desse operador ótimo. O chamado fenômeno
da curva em U [Campos, 2001] explica que uma janela pequena cobre uma parte
muito pequena do espaço, enquanto uma janela muito grande aumenta o erro
de estimação, estando ambos extremos associados a maiores erros.
Portanto, existe um tamanho de janela ótimo. A figura 5 é a curva
em U (gráfico tamanho da janela X erro do operador) que demostra a existência
de um tamanho de janela ótimo.
 |
|
Figura 5
|
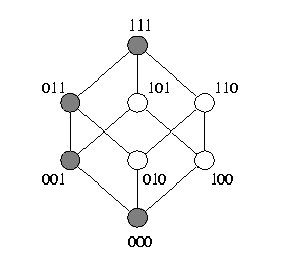
O conceito de reticulado booleano foi aplicada à idéia de janela, cada janela correspondendo a um ponto no reticulado. Por exemplo, no reticulado da figura 6, os pontos {001, 010, 100} correspondem a 3 diferentes janelas de tamanho 1, {011, 101, 110} correspondem a 3 janelas de tamanho 2 e assim por diante. Inicialmente, treinamos o operador com a janela "0" e avaliamos o erro.
 |
|
Figura 6
|
Seguindo um caminho ascendente no reticulado (por exemplo o caminho marcado com pontos escuros), treinamos o operador com uma janela de tamanho 1 e avaliamos o erro. Seguindo esse caminho ascendente no reticulado, o tamanho da janela aumenta progressivamente e o erro tende a diminuir até que a janela produza um operador que gere um erro mínimo. Quando o erro avaliado for maior que o do operador anterior, acabamos de avançar sobre o operador ótimo para esse caminho. Se o operador treinado com a janela 011 gerasse um erro maior do que o gerado pela janela 001 então 001 seria a janela ótima para o caminho marcado com pontos escuros. Essa janela ótima para esse caminho pode não ser ótima global, portanto temos que continuar avaliando o erro dos operadores treinados com janelas de outros caminhos. No entanto todos os pontos do caminho "acima" de 001 (XX1) não precisam mais ser testados.
Essa técnica de busca exaustiva diminui bastante a tempo para encontrar o operador ótimo mas não o suficiente para torná-la viável. Isso devido à complexidade do processo de treinamento de operadores.
3. Conclusão
A realização desse projeto de iniciação científica, além de contribuir com o grupo de pesquisa BIOINFO-USP, aproximou o aluno à área de reconhecimento de padrões e bioinformática. Proporcionando-o a oportunidade de experimentar um trabalho de pesquisa, buscando informações em livros, artigos científicos e na rede mundial. A implementação dos algoritmos estudados consolidou os conhecimentos adquiridos, assim como a aplicação em problemas de bioinformática pôs a prova o trabalho realizado e permitiu que fosse feito um trabalho de avaliação dos resultados.
4. Relação com o curso BCC
Durante o período de pesquisa, foi consultado diversos livros, e artigos relacionados ao tema reconhecimento de padrões. Como a área de reconhecimento de padrões é tem em sua essência a estatística e a álgebra, foi fundamental para a compreensão dessa literatura técnica os conhecimentos básicos adquiridos nas disciplinas de estatística, álgebra linear e álgebra booleana. Além dessas, as disciplinas de Análise e Reconhecimento de Formas e Aprendizado Computacional contribuíram para a fixação dos conhecimento adquiridos durante o período de pesquisas, pois abortaram exatamente os algoritmos estudados nesse projeto de iniciação científica. A disciplina de Estrutura de Dados foi de grande importância para a fase de implementação do projeto.
A realização desse projeto de iniciação científica mostrou-me que o IME prepara excepcionalmente bem seus alunos para seguir carreira acadêmica. O aluno que seguiu o curso com seriedade é capaz de realizar, discutir e até mesmo inovar a ciência.
Dentre os desafios desse projeto vale ressaltar a necessidade de compreender artigos científicos tanto da área da ciência da computação quanto os artigos da biologia que fizeram parte do contexto do projeto. Desafio superado graças ao preparo adquirido no curso BCC.
Referências Bibliográficas
Duda, R.O. and Hart, P.E., Pattern classification and scene analysis. John iley Sons, New York, 1973.
Young, T. Y., and Calvert, T.W., Classification, estimation and pattern recognition. Elsevier, New York, 1974.
Gose, E.; Johnsonbaugh, R.; and S. Jost, Pattern Recognition and Image Analysis, Prentice Hall, NJ, 1996.
Setubal, J.; Meidanis, J., Introduction to Computational Molecular Biology. Pws Publishing Company, 1997.
Jain, A. K.; Murty, M. N. and Flynn, P. J., Data Clustering: A Review. ACM Computing Surveys, 31, Setembro 1999
Brown, P.O.; and Botstein, D., Exploring the new world of the genome with DNA microarrays. Nature Genetics, 21, pp 33-37, Jan 1999.
Eisen, M.B.; Spellman, P.T.; Brown, P.O. and Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95, 14863-14868, Dec 1998.
Tibshirani , R.; Hastir, T.; Eisen M.B.; Ross, D.; Botstein, D. and Brown, P. Clustering methods for the analysis of DNA microarray data. Department of Health Research and Policy, Department of Biochemistry, Stanford Universty. October 15, 1999
Dougherty, E.; Bittner, M.; Meltzer P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; Sampas, N.; Wang, E.; Marincola, F.; Gooden, C.; Lueders, J.; Glatfelter, A.; Pollock, P.; Carpten, J.; Gillanders, E.; Leja, D.; Dietrich, K.; Beaudry, C.; Berens, M.; Alberts, D.; Sondak, V.; Hayward, N.; and Trent, J., Molecular classification of cutaneous malignant melanoma by gene expression profiling, Nature 406, pp 536 – 540, 2000
Bezdek, J.C. Pattern Recognition with Fuzzy Objective Function Algorithms. Plenum Press, New York, 1981
Press, W.H., Teukolsky, S.A., Vetterling, W.T., Flannery, B.P. Numerical Recipes: The art of scientific computing. Cambridge University Press, 1992. http://www.nr.com
Cesar Jr., R. M.; Costa, L. F. Shape Analysis and Classification: Theory and Pratice CRC Pess, 2001
Campos, T.E. Técnicas de Seleção de Características com Aplicações em Reconhecimento de Faces Dissertação de Mestrado, DCC-IME-USP, 2001
Barrera, J.; Hirata, N.S.T; Dougherty, E.R. Iterative Design of Morphological Binary Image Operators Optical Engineering, vol 39 No. 12, December 2000
Dougherty, E.R.; Barrera, J.; Hirata, N.S.T A Swiching Algorithm for Design of Optimal Increasing Binary Filter Over Large Windows Pattern Recognition 33 (2000) 1059-1081
Anthony, M.; Biggs, N. Computational Learning Theory An Introduction Cambridge University Press, 1992
Dougherty, E.; Bittner, M.; Meltzer P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; Sampas, N.; Wang, E.; Marincola, F.; Gooden, C.; Lueders, J.; Glatfelter, A.; Pollock, P.; Carpten, J.; Gillanders, E.; Leja, D.; Dietrich, K.; Beaudry, C.; Berens, M.; Alberts, D.; Sondak, V.; Hayward, N.; and Trent, J., Molecular classification of cutaneous malignant melanoma by gene expression profiling, Nature 406, pp 536 – 540, 2000
NHGRI Microarray Project Image Analysis http://www.nhgri.nih.gov/DIR/LCG/15K/HTML/img_analysis.html