Next: Desenvolvimento Up: Descrição Previous: Sobre análise genômica
Como base de testes foi selecionado um conjunto de 26 Báculo vírus cuja árvore filogenética é conhecida.
Os genomas são submetidos primeiramente ao programa getorf, que extrai os segmentos mais significativos das seqüências genéticas. Esses dados são então submetidos ao Blast, que compara cada seqüência S com as demais, listando as que têm mais semelhanças com S.
Em uma outra etapa, os arquivos gerados pelo Blast são processados e deles são extraídas somente as informações relevantes para o BlastPhen: os valores dos bit scores, raw scores, identities e positives. Também nesta etapa são calculadas as médias, medianas e modas dos atributos citados para cada comparação entre seqüências.
Tendo reunido os dados necessários, o BlastPhen calcula as distâncias entre as espécies de acordo com as métricas abaixo:
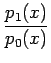 dx
dx
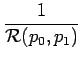
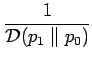 +
+ 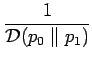
Como podemos verificar, tais medidas de distâncias referem-se a dados contínuos enquanto que neste projeto estamos lidando com dados discretos. Portanto, com a finalidade de adaptar as informações, estão sendo utilizados histogramas para agrupá-las.
Dois problemas surgiram com a utilização dos histogramas: quantas
classes devem ser utilizadas para agrupar os dados e o que fazer
quando não há dados em uma classe com relação a uma função e com
relação a outra há dados (i.e.
p0(x) = 0 e
p1(x) ![]() 0 e
vice-versa).
0 e
vice-versa).
Os resultados são então organizados em tabelas, facilitando a consulta e a construção da árvore filogenética.
Duas grandes dificuldades estão sendo as escolhas do tamanho do orf utilizado pelo getorf e da matriz de substituição de aminoácidos aplicada pelo Blast.
Após o cálculo das distâncias entre as espécies, o BlastPhen agrupa as mesmas em tabelas de tal forma que cada tabela consiste em um bonsai. Se pensarmos em cada espécie como sendo um vértice de um grafo, cada bonsai representa um clique desse grafo.
Ricardo Nishikido Pereira 2004-12-06